Welcome to the Weihl Lab!
Our goal is to understand the molecular mechanisms of protein inclusion formation, disaggregation and clearance in myodegenerative and neurodegenerative diseases.

Muscle-specific inactivation of VCP leads to necrotic myopathy with autophagic pathology. 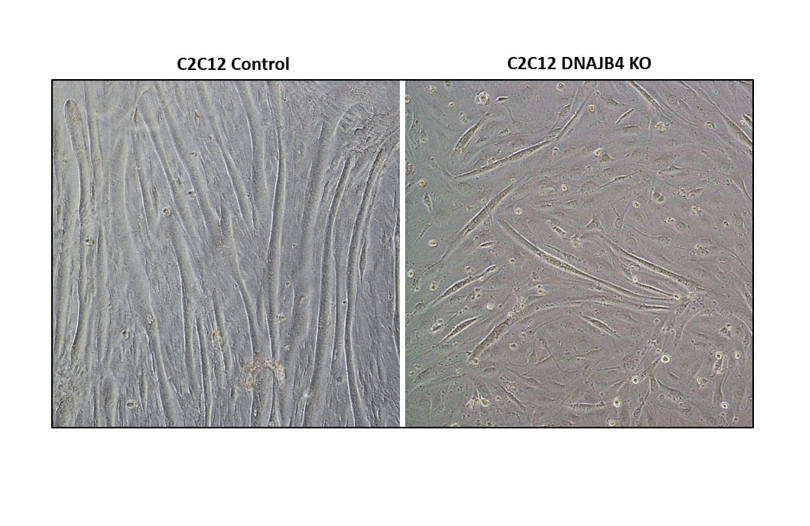
WT & DNAJB4 knockout mouse myotubes associated with a dominantly inherited distal myopathy. 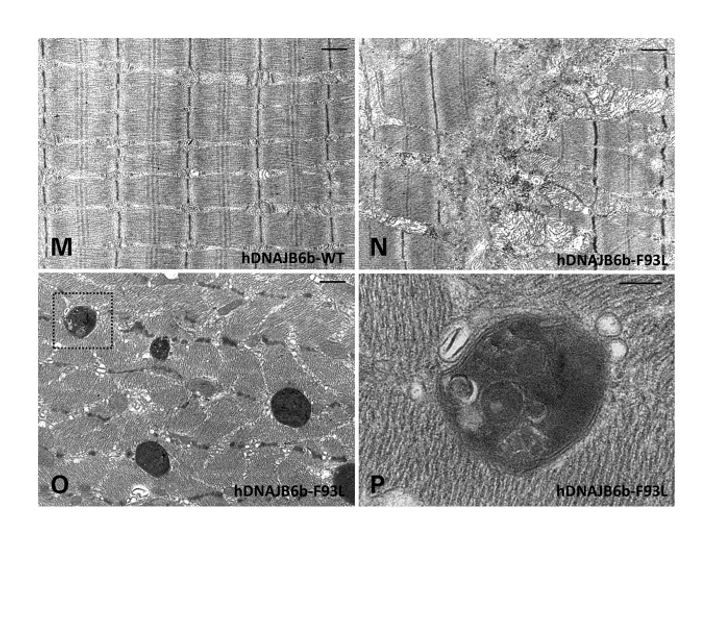
Electron micrographs of TA muscle from WT & F93L knock-in mice associated with LGMD-1D. 
Deep mutational scanning of SCGB provides functional evidence to predict missense variant pathogenicity in LGMD-R4/2E. 
VCP associated IBM, PDB, FTD, and ALS.
We believe that dysfunction in protein quality control (molecular chaperones) and protein degradation pathways (e.g. the ubiquitin proteasome system, autophagy and endocytosis) disrupt cellular protein homeostasis resulting in degenerative phenotypes and normal aging. We employ complementary experimental techniques such as biochemistry, cell culture, animal models, and patient tissue to investigate these processes.
Our lab is particularly interested in the pathogenesis of inclusion body myopathy with Paget’s disease of the bone and frontotemporal dementia (IBMPFD) and limb-girdle muscular dystrophy (LGMD). IBMPFD is a multisystem degenerative disorder caused by missense mutations in p97/VCP/cdc48. IBMPFD muscle and brain tissue contains ubiquitinated protein inclusions associated with cellular degeneration and vacuolation. LGMDs are a group of muscular diseases characterized by predominant proximal muscle weakness. LGMD subtypes are highly variable in terms of severity, age of onset, and speed of disease progression and do not share a common pathological mechanism. However, many forms of LGMD involve disruptions in normal protein quality control processes, like protein folding, assembly, transport and degradation, which can impair normal cellular functions and ultimately lead to cell death.
Positions
The Weihl Lab is looking for motivated research assistants, undergraduate students, graduate students and postdocs. If interested in working with us or if you have any questions, please contact us.
Featured Publications
Seeding-competent TDP-43 persists in human, patient, and mouse muscle (Sci. Transl. Med.,) 2024
Genotype-phenotype correlation in recessive DNAJB4 myopathy (Acta Neuropathol. Commun., 2024 Pre-print)
Genetic deletion of skeletal muscle iPLA2γ results in mitochondrial dysfunction, muscle atrophy and alterations in whole-body energy metabolism (iScience, 2023)
DNAJB6 isoform specific knockdown: Therapeutic potential for limb girdle muscular dystrophy D1 (Molecular Therapy – Nucleic Acids, 2023)
Distinctive chaperonopathy in skeletal muscle associated with the dominant variant in DNAJB4 (Acta Neuropathol., 2023)
Loss of function variants in DNAJB4 cause a myopathy with early respiratory failure (Acta Neuropathol., 2023)
VCP suppresses proteopathic seeding in neurons (BMC, 2022)
Molecular and cellular basis of genetically inherited skeletal muscle disorders (Nature Reviews, 2021)
Inhibition of DNAJ-HSP70 interaction improves strength in muscular dystrophy (JCI, 2020)
Desmin forms toxic, seeding-competent amyloid aggregates that persist in muscle fibers (PNAS, 2019)
Keap1/Cullin3 Modulates p62/SQSTM1 Activity via UBA Domain Ubiquitination (CellPress, 2017)





