Inclusion body myopathy, paget’s disease of the bone and frontotemporal dementia or IBMPFD, is a multisystem degenerative disorder due to missense mutations in the multifunctional AAA+ ATPase, p97/VCP/cdc48. IBMPFD is autosomal dominantly inherited disorder and has three variably penetrant phenotypes: inclusion body myopathy (IBM), paget’s disease of the bone (PDB), and frontotemporal dementia (FTD). Other phenotypes have been described including cardiomyopathy, cataracts and neuropathy.
Clincial Phenotypes in IBMPFD
IBM is characterized by progressive debilitating weakness affecting both the upper and lower extremities. Patients can present with syndromes mimicking limb-girdle muscular dystrophy, fascioscapulohumeral dystrophy or a distal myopathy.
PDB is a progressive bone disorder due to excessive breakdown and formation of bone tissue resulting in characteristic lytic lesions on autoradiography. This causes bone to weaken, resulting in bone pain, arthritis, deformities, and fractures.
FTD is the second most common cause of senile dementia. Unlike Alzheimer’s dementia, FTD patients have preserved working memory early in the disease course and instead have personality changes such as disinhibition along with difficulties in speech production, attention, reasoning and language.
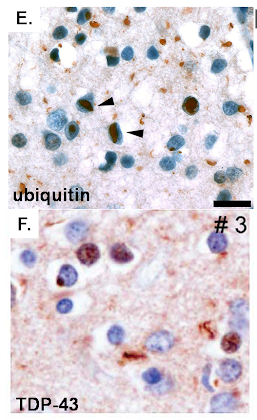
Pathologic features of IBMPFD
Skeletal muscle contains several characteristic features consistent with IBM including (A) rimmed vacuoles and (B) intranuclear and sarcoplasmic ubiquitinated protein inclusions. In additon, we have identified (C) intranuclear and sarcoplasmic VCP and (D) sarcoplasmic TDP-43 positive inclusions.
Affected neurons in the frontal cortex are tau negative. Instead they have intranuclear and cytoplasmic ubiquitinated and TDP-43 positive inclusions. In addition rare VCP positive intranuclear inclusions are present.